MASLD Drug Discovery: Why the Cell Model You Choose Matters More Than You Think
Metabolic dysfunction-associated steatotic liver disease has become one of the most intensely pursued therapeutic areas in drug development. The scale of the unmet need is substantial: MASLD affects an estimated 25 to 30 percent of the global adult population, and its more severe form, metabolic dysfunction-associated steatohepatitis, is projected to become the leading cause of liver transplantation in many countries within the next decade. Despite this, the history of MASLD drug development is largely one of late-stage failures. Understanding why requires looking closely at the preclinical models used to advance candidates to the clinic — and what those models can and cannot tell you.
What MASLD Is and Why It Is Hard to Model
MASLD is not a single disease state but a spectrum of conditions ranging from simple hepatic steatosis — fat accumulation in liver cells without significant inflammation or fibrosis — through progressive steatohepatitis with hepatocellular injury and inflammation, to advanced fibrosis and cirrhosis. The clinical trajectory is heterogeneous: most patients with steatosis will not progress to steatohepatitis, but a subset will progress rapidly to fibrosis and liver failure, and identifying which patients are on that trajectory before the damage is advanced is one of the central challenges in clinical MASLD management.
This heterogeneity creates the first modelling challenge. A preclinical model designed to capture simple steatosis will not capture the inflammatory and fibrogenic mechanisms that drive disease progression. A model designed to capture advanced fibrosis may not reflect the earlier stages of disease where most therapeutic intervention is directed. And a model that does not account for the genetic risk factors that determine which patients progress — PNPLA3 I148M, TM6SF2 E167K, MBOAT7 variants, and others — may miss drug effects that are specific to genetically susceptible patient subpopulations.
The Limitations of Standard Animal Models
Diet-induced and genetic mouse models of MASLD have been central to preclinical drug development for decades. The most widely used include the high-fat diet model, the methionine-choline-deficient diet model, the fructose-enriched diet model, and genetic models such as the ob/ob mouse.
Each captures some aspects of human MASLD biology and misses others, often substantially. The methionine-choline-deficient diet model produces rapid steatohepatitis and fibrosis but does so through a mechanism involving nutritional deficiency rather than metabolic excess, and is accompanied by weight loss rather than the weight gain and insulin resistance that characterise human disease. The high-fat diet model reproduces obesity and insulin resistance more faithfully but produces only modest hepatic inflammation and minimal fibrosis in most strains. Genetic models such as ob/ob develop severe obesity and steatosis but require additional insults to develop significant steatohepatitis.
The broader problem is species biology. Mice metabolise lipids differently from humans, have different compositions of hepatic fatty acid pools, different lipoprotein biology, and different susceptibilities to hepatic fibrogenesis. Drugs that show efficacy in mouse MASLD models have a substantially worse translation record into human clinical trials than most therapeutic areas. Teufel et al. (2016) performed a systematic transcriptome-wide analysis comparing nine mouse MASLD models against human liver tissue, finding that differences between human and mouse transcriptomes were significantly larger than differences between disease stages or models — a fundamental challenge for preclinical translation.
What In Vitro Models Are Currently Used and Where They Fall Short
Standard in vitro MASLD modelling uses hepatocyte cell lines or primary hepatocytes exposed to fatty acids, glucose, fructose, or other lipotoxic stimuli to induce steatosis in culture.
HepG2 cells are widely used for steatosis modelling because they accumulate lipid droplets readily in response to oleic acid or palmitate treatment, and lipid accumulation is easy to quantify by fluorescent staining. The problem is that HepG2 cells lack the metabolic competence of primary hepatocytes, the inflammatory signalling that characterises MASH, the stellate cell and immune cell interactions that drive fibrogenesis, and the genetic background of any specific patient population. They model lipid accumulation in a liver cancer cell line, which is not the same as modelling MASLD in a human hepatocyte.
Primary human hepatocytes offer better metabolic competence but suffer from the donor variability and dedifferentiation problems discussed elsewhere. They are also not amenable to the long-term culture required to model the slow progression of MASLD from steatosis to steatohepatitis to fibrosis: by the time the culture has dedifferentiated, the biology of interest has changed.
Co-culture systems that combine hepatocytes with hepatic stellate cells and Kupffer cells can model aspects of the inflammatory and fibrogenic crosstalk that drives disease progression beyond simple steatosis. These systems are more informative but harder to standardise and more difficult to scale to drug discovery throughput.
What iPSC-Derived Models Add
iPSC-derived hepatocytes bring several specific advantages to MASLD drug discovery that conventional models cannot match.
Genetic risk variant modelling. The most important genetic risk factors for MASLD progression — PNPLA3 I148M and TM6SF2 E167K — can be introduced into iPSC lines by CRISPR editing, generating isogenic pairs that differ only at the variant of interest. This enables the direct study of how these genetic variants affect lipid metabolism, inflammatory signalling, and drug response, without the confounding effects of different genetic backgrounds.
Conversely, iPSC lines can be derived from patients carrying these variants naturally, preserving the full polygenic context of disease-associated genetic backgrounds in a way that isogenic CRISPR models cannot.
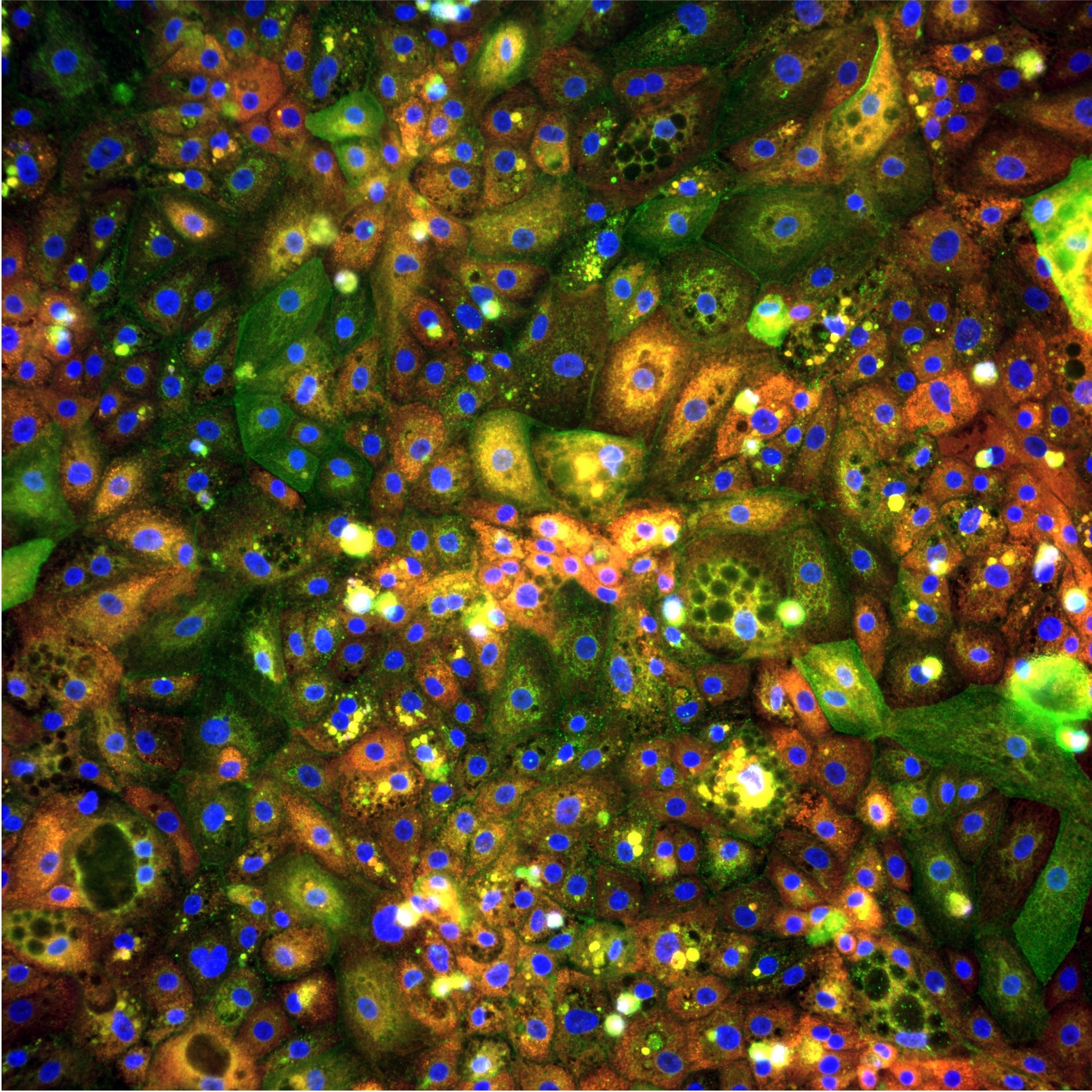
Inducible disease phenotype. pixlbio's MASLD iPSC hepatocyte models recapitulate steatosis, insulin resistance, and inflammatory signatures in response to standardised lipotoxic stimuli. Increased lipid accumulation, validated by BODIPY staining, is accompanied by reduced AKT phosphorylation following insulin stimulation, confirming the development of hepatic insulin resistance, and elevated expression of pro-inflammatory cytokines including IL-1β, IL-6, IL-8, and TNFα. These are not simply fat-loaded cells. They are cells that have adopted a disease-relevant functional state that can be quantified and used as a screening readout. These findings are further supported by work presented by the pixlbio team at the Society for Endocrinology BES 2025, where iPSC-derived hepatocytes were shown to recapitulate the full MASLD phenotype in vitro across both wild-type and PNPLA3-I148M risk-variant lines (Endocrine Abstracts, 2025. DOI: 10.1530/endoabs.109.OC6.5).
Long-term culture stability. Unlike primary hepatocytes, iPSC-derived hepatocytes maintain their phenotype over the extended culture periods required for MASLD modelling. This enables repeated dosing studies, disease progression experiments, and the kind of chronic compound exposure that is relevant to the clinical treatment context.
Morphological profiling. Combined with Cell Painting, iPSC-derived MASLD hepatocytes generate rich morphological signatures of disease state and drug response. Changes in lipid droplet morphology, mitochondrial organisation, nuclear shape, and cytoskeletal architecture can all be quantified at the single-cell level across thousands of cells per well. This morphological readout captures aspects of hepatocyte biology that biochemical and viability assays miss entirely, and may be sensitive to compound effects that occur at concentrations below those required to produce a measurable change in any single biochemical marker.
Co-culture compatibility. Our MASLD hepatocyte models are compatible with co-culture with pixStellate hepatic stellate cells, enabling the modelling of hepatocyte-stellate crosstalk and the fibrogenic progression from steatohepatitis to early fibrosis. This moves the model beyond simple steatosis towards the disease biology that is most relevant to therapeutic intervention.
What Is Still Missing
Intellectual honesty requires acknowledging what current iPSC-based MASLD models do not yet capture.
The immune component of steatohepatitis — specifically the role of Kupffer cells, hepatic macrophages, and circulating immune cells in driving and amplifying hepatic inflammation — is not present in hepatocyte-only or hepatocyte-stellate co-culture models. Kupffer cell activation is central to MASH pathophysiology, and drugs targeting macrophage polarisation or inflammatory signalling pathways in Kupffer cells will not be evaluated accurately in models that lack them.
The metabolic context of MASLD — the whole-body insulin resistance, adipose tissue dysfunction, and gut-liver axis alterations that characterise the disease in patients — is also absent from cell-based models. A drug that addresses systemic metabolic dysfunction rather than hepatocyte-intrinsic disease mechanisms may show little or no effect in a hepatocyte model while being highly effective in patients.
And the slow, multi-decade natural history of MASLD cannot be fully recapitulated in any in vitro system with current culture technologies. What can be modelled is the cellular and molecular biology of specific disease stages, not the full disease trajectory.
These limitations do not invalidate iPSC-based MASLD models as drug discovery tools. They define the questions those models can and cannot answer, and understanding that boundary is essential for using them well.
The Drug Development Landscape
Several therapeutic approaches have shown clinical signal in MASLD and MASH trials, and the mechanistic diversity of the clinical pipeline reflects the complexity of the disease. Resmetirom, a thyroid hormone receptor-β agonist, became the first FDA-approved treatment for MASH with fibrosis in 2024, following the pivotal MAESTRO-NASH phase 3 trial demonstrating significant improvements in both NASH resolution and fibrosis stage reduction. GLP-1 receptor agonists, including semaglutide, have shown significant reductions in MASH histology in clinical trials. FXR agonists, ACC inhibitors, and ASK1 inhibitors have all been in clinical development with varying results.
The mechanistic heterogeneity of these approaches — from lipid metabolism to inflammatory signalling to fibrogenesis to hormonal regulation — reinforces the point that no single preclinical model will be informative for all of them. A good MASLD drug discovery programme uses models matched to the mechanism being targeted, rather than applying a single generic steatosis model across all programmes.
References
pixlbio's MASLD iPSC hepatocyte models recapitulate steatosis, insulin resistance, and inflammatory signatures from defined donor backgrounds including PNPLA3 and TM6SF2 risk variants. Our pixCellPaint platform enables morphological profiling of disease state and drug response at scale.
To discuss MASLD modelling for your programme, book a call with our team.
Tags: MASLD · MASH · NAFLD · Liver Disease · Drug Discovery · iPSC Hepatocytes · Steatosis · Fibrosis · PNPLA3 · TM6SF2 · Cell Painting · Hepatotoxicity
Conclusion
- Teufel A et al. (2016). Comparison of gene expression patterns between mouse models of nonalcoholic fatty liver disease and liver tissues from patients. Gastroenterology, 151(3):513–525. https://doi.org/10.1053/j.gastro.2016.05.051
- Friedman SL et al. (2018). Mechanisms of NAFLD development and therapeutic strategies. Nature Medicine, 24:908–922. https://doi.org/10.1038/s41591-018-0104-9
- Younossi ZM et al. (2016). Global epidemiology of nonalcoholic fatty liver disease: meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology, 64(1):73–84. https://doi.org/10.1002/hep.28431
- Romeo S et al. (2008). Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nature Genetics, 40(12):1461–1465. https://doi.org/10.1038/ng.257
- Kozlitina J et al. (2014). Exome-wide association study identifies a TM6SF2 variant that confers susceptibility to nonalcoholic fatty liver disease. Nature Genetics, 46(4):352–356. https://doi.org/10.1038/ng.2901
- Tacke F (2017). Targeting hepatic macrophages to treat liver diseases. Journal of Hepatology, 66(6):1300–1312. https://doi.org/10.1016/j.jhep.2017.02.026
- Harrison SA et al.; MAESTRO-NASH Investigators (2024). A phase 3, randomized, controlled trial of resmetirom in NASH with liver fibrosis. New England Journal of Medicine, 390(6):497–509. https://doi.org/10.1056/NEJMoa2309000
- pixlbio / DefiniGEN team (2025). iPSC-derived hepatocytes offer a novel platform for modelling metabolic dysfunction-associated steatotic liver disease (MASLD) in vitro. Endocrine Abstracts, 109 OC6.5. https://doi.org/10.1530/endoabs.109.OC6.5